Getting started with the pedsuite
pedsuite.RmdInstallation
The following command installs the latest official versions of the pedsuite packages:
install.packages("pedsuite")Alternatively, you can install the development versions from GitHub:
# install.packages("devtools")
devtools::install_github("magnusdv/pedsuite")If you only need a few of the packages, you may choose to install them individually instead of the entire collection.
A quick tour
The aim of this vignette is to illustrate a few of the possibilities of the pedsuite packages. It should be noted that we are barely scratching the surface here; in particular several packages are not even mentioned. For a more comprehensive overview, I recommend the book.
To get started we load the pedsuite package, which is a convenient shortcut for loading all the core packages, making their methods available in the current R session.
library(pedsuite)
#> Loading required package: forrel
#> Loading required package: pedtools
#> Loading required package: pedprobr
#> Loading required package: ribd
#> Loading required package: verbalisr1. Create a pedigree (pedtools)
We begin by creating and plotting a pedigree with a child of first cousins:
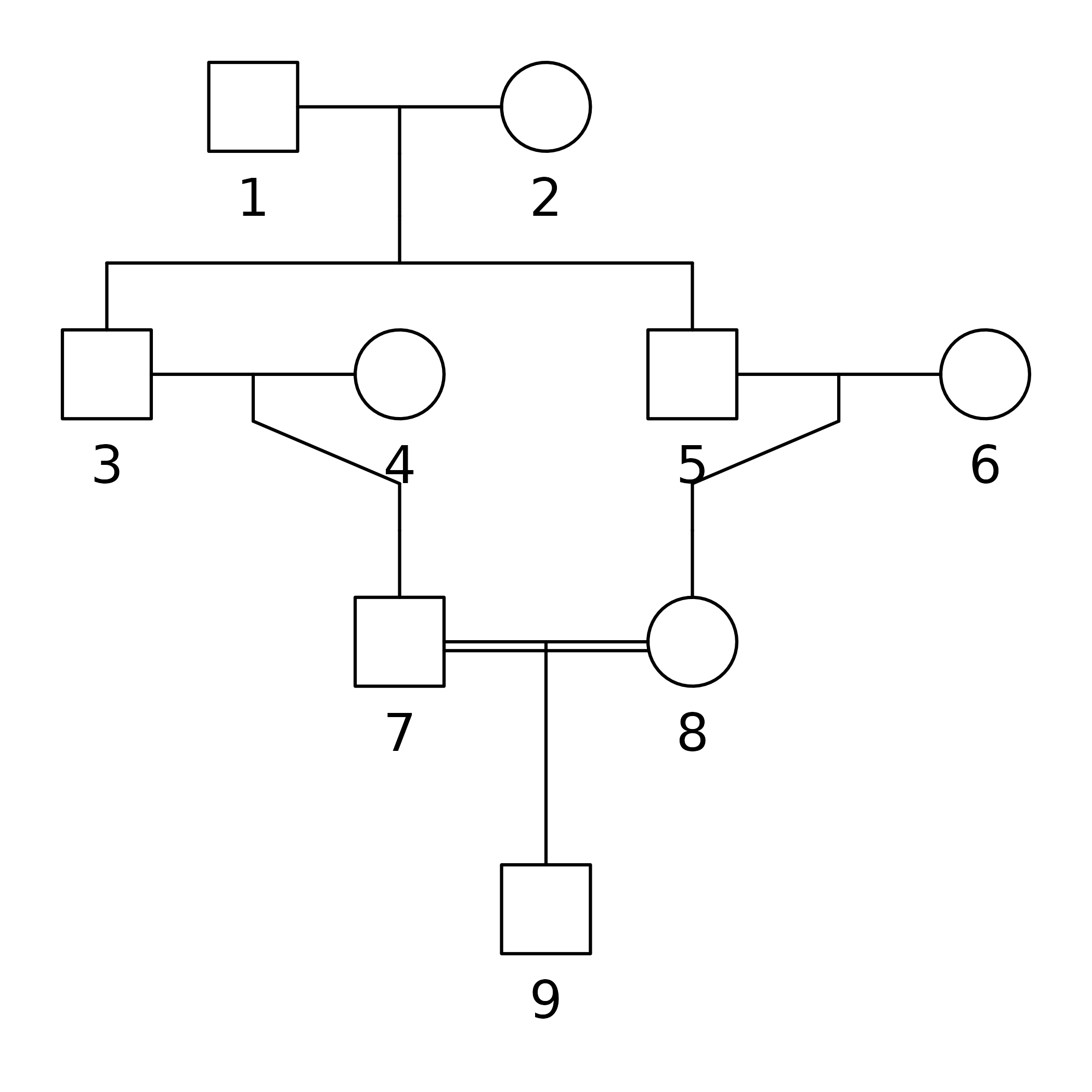
For symmetry let us change the sex of individual 3. We also take the
opportunity to showcase some of the plot options (many more are
available - see the help page by typing ?plotmethods):
x = swapSex(x, ids = 3)
#> Changing sex of spouses as well: 4
plot(x,
hatched = 9,
carrier = 7:8,
fill = list(pink = 1),
textAnnot = list(inside = c("1" = "?")))
2. Calculate the inbreeding coefficient (ribd)
The inbreeding coefficient of a pedigree member is defined as the probability of autozygosity at a random autosomal locus. That is, the probability that the two homologous alleles have the same origin within the pedigree.
For a child of first cousins one can work out by pen and paper that
.
Alternatively, we can calculate it with the ribd
function inbreeding().
inbreeding(x, ids = 9)
#> [1] 0.0625The output agrees with .
3. Realised inbreeding (ibdsim2)
For any particular child of first cousins, the actual autozygous fraction of the genome (except X & Y) is called the coefficient of realised inbreeding, denoted . This may deviate substantially from the pedigree-based expectation .
We can simulate the distribution of with the ibdsim2 package. Since this is not a core package we must load it separately.
First, we use the function ibdsim() to simulate the
recombination process in the entire pedigree, 200 times:
sims = ibdsim(x, N = 200, seed = 123)
#> Simulation parameters:
#> Simulations : 200
#> Chromosomes : 1-22
#> Genome length: 2875 Mb
#> 2602.29 cM (male)
#> 4180.42 cM (female)
#> Recomb model : chi
#> Target indivs: 1-9
#> Skip recomb : -
#> Total time used: 1.87 secsNow extract the autozygous segments of each simulation.
fr = realisedInbreeding(sims, id = 9)Here is a summary of the first 6 simulations, including the number of segments and various length statistics:
head(fr$perSimulation)
#> nSeg meanLen totLen maxLen minLen fReal
#> 1 11 19.04942 209.5437 37.56569 0.8072431 0.06178760
#> 2 12 14.17475 170.0970 48.43677 1.3413298 0.05015605
#> 3 12 12.87744 154.5293 26.11588 0.1475564 0.04556565
#> 4 19 17.43950 331.3506 58.64604 1.2084099 0.09770450
#> 5 16 16.96611 271.4577 44.94334 1.0386058 0.08004404
#> 6 13 17.84722 232.0139 49.91368 0.9532618 0.06841334And here is a histogram of the realised inbreeding coefficients (given in the right-most column above):
hist(fr$perSimulation$fReal, xlim = c(0, 0.15), breaks = 16, xlab = "f_R", main = NULL)
# Expected value
abline(v = 1/16, col = 2)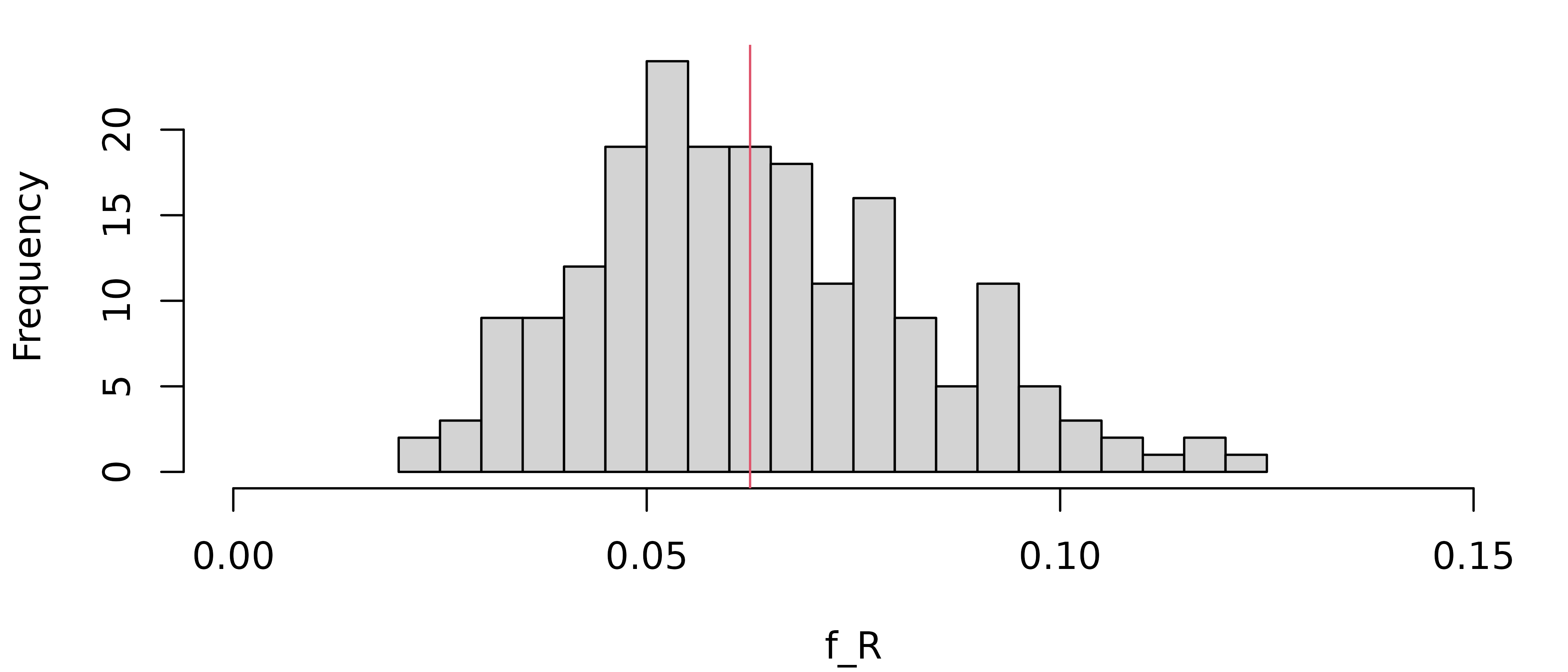
As we see, the distribution centres around the expectation
(red vertical line) but has substantial spread. The sample standard
deviation can be found in fr$stDev, which in our case is
0.02.
4. Marker simulation (forrel)
Note that everything we have done so far has been purely theoretical, with no markers involved. In medical and forensic applications we usually work with genetic data in the form of marker genotypes, so let us simulate such a dataset for our family.
The markerSim() function of the forrel
package simulates the genotypes of pedigree members for a specific type
of markers. For instance, here we produce 500 SNPs with alleles
A and B (equally frequent, by default):
y = markerSim(x, N = 500, alleles = c("A", "B"))
#> Unconditional simulation of 500 autosomal markers.
#> Individuals: 1, 2, 3, 4, 5, 6, 7, 8, 9
#> Allele frequencies:
#> A B
#> 0.5 0.5
#> Mutation model: No
#>
#> Simulation finished.
#> Calls to `likelihood()`: 0.
#> Total time used: 0 seconds.We can see the genotypes of the first few markers by printing
y to the console.
y
#> id fid mid sex <1> <2> <3> <4> <5>
#> 1 * * 1 A/B B/B A/A B/B A/A
#> 2 * * 2 A/B A/B A/B A/A A/B
#> 3 1 2 2 A/B A/B A/B A/B A/B
#> 4 * * 1 A/B A/B B/B A/B A/B
#> 5 1 2 1 B/B A/B A/B A/B A/A
#> 6 * * 2 A/B B/B A/A A/B A/A
#> 7 4 3 1 B/B A/A B/B A/B A/B
#> 8 5 6 2 A/B A/B A/B B/B A/A
#> 9 7 8 1 B/B A/A B/B B/B A/A
#> Only 5 (out of 500) markers are shown.5. Inference of pairwise relationships (forrel)
If this was a real dataset, a natural quality control step would be
to check correctness of the pedigree. One way to do this is to use the
data to estimate each pairwise relationship, and compare the result with
the pedigree. The function checkPairwise() does all of
this, and presents the result in a relationship triangle.
checkPairwise(y)
#> Excluding inbred individuals: 9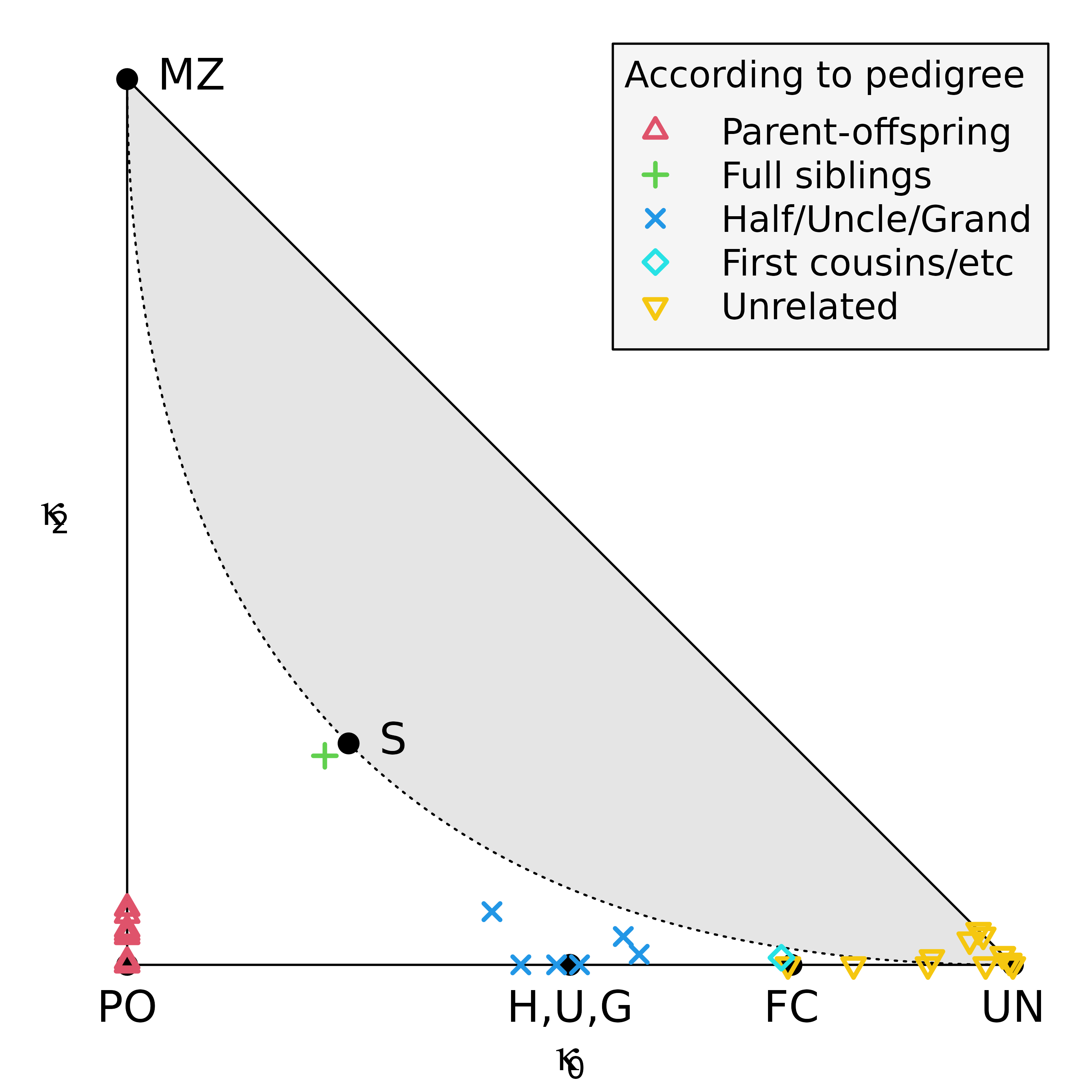
The plot shows that all pairwise estimates are near their expected location in the triangle.
Where to go from here
If you enjoyed this quick tour and would like more details, you should check out the GitHub README files of the packages that interest you, for instance:
- pedtools: Creating and working with pedigrees and marker data
- forrel: Forensic pedigree analysis and relatedness analysis
- ribd: Computation of pedigree-based relatedness coefficients
- verbalisr: Verbal descriptions of pedigree relationships
- ibdsim2: Simulation of identity-by-descent sharing by family members
- dvir: Disaster victim identification
Try the interactive pedigree builder QuickPed: https://magnusdv.shinyapps.io/quickped.
The pedtools vignette contains details on pedigree objects in R and how to plot and manipulate them.